

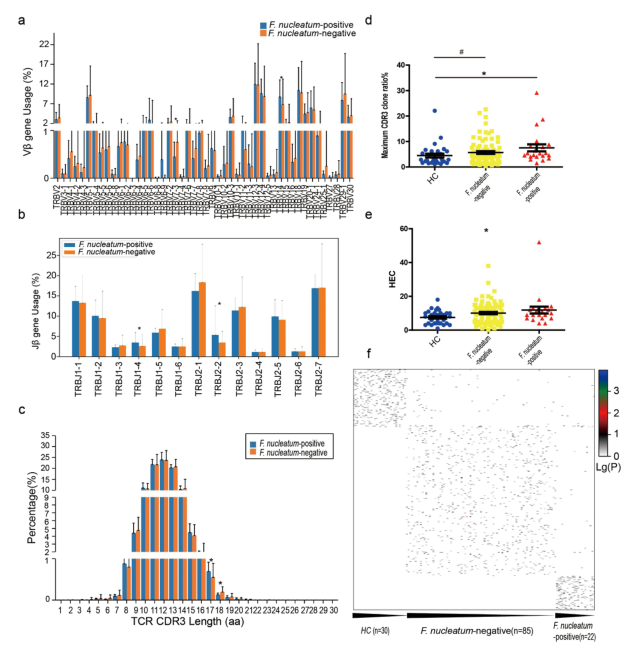
зїУзЫіиВ†зЩМзЫЄеЕ≥зЪДTзїЖиГЮеПЧдљУеЇУеЉВеЄЄдЄОиВ†йБУеЊЃзФЯзЙ©зїДиљђзІїеТМдљУзїЖиГЮз™БеПШжЬЙеЕ≥ еПСи°®жЬЯеИКпЉЪGut microbes еПСи°®жЧґйЧіпЉЪ2023еєі10жЬИ ељ±еУНеЫ†е≠РпЉЪ12.2 з†Фз©ґжЦєж≥ХпЉЪ вС†жФґйЫЖдЇЖ107еРНзїУзЫіиВ†зЩМжВ£иАЕеТМ30еРНеБ•еЇЈеѓєзЕІиАЕзЪДи°Ажґ≤ж†ЈжЬђињЫи°МTCRжµЛеЇПгАВвС°жФґйЫЖдЇЖ97еРНзїУзЫіиВ†зЩМжВ£иАЕеТМ30еРНеБ•еЇЈеѓєзЕІиАЕзЪДз≤™дЊњж†ЈжЬђињЫи°МеЃПеЯЇеЫ†зїДе≠¶з†Фз©ґгАВвСҐеѓє79дЊЛзїУзЫіиВ†зЩМжВ£иАЕзЪДиВњзШ§зїДзїЗеТМйВїињСзїДзїЗињЫи°МдЇЖйЭґе§ЦжШЊе≠РзїДжµЛеЇПгАВвС£зїЯиЃ°еИЖжЮР з†Фз©ґиГМжЩѓ ¬† ¬† ¬† зїУзЫіиВ†зЩМ(CRC)жШѓеЕ®зРГжЬАеЄЄиѓКжЦ≠зЪДдЄЙе§ІзЩМзЧЗдєЛдЄАгАВзДґиАМињЩзІНеЕ®зРГжАІжБґжАІиВњзШ§еПСзФЯеТМеПСе±ХзЪДжљЬеЬ®жЬЇеИґе∞ЪжЬ™еЃМеЕ®йШРжШОгАВдЇЇз±їиВ†йБУдЄ≠е≠ШеЬ®жЬЙињС1.5еЕђжЦ§зЪДеЊЃзФЯзЙ©зЊ§пЉМжЯРдЇЫзЧЕжѓТеТМ/жИЦзїЖиПМеПѓиГљдЄОдЇЇз±їзЩМзЧЗжЬЙеЕ≥гАВзїУзЫіиВ†зЩМзЪДеПСзФЯеПСе±ХдЄОеЉВеЄЄзЪДеЕНзЦЂеЊЃзОѓеҐГзЫіжО•зЫЄеЕ≥пЉМжЈЛеЈізїЖиГЮеЇУеПѓдї•йАЪињЗдЇТи°•еЖ≥еЃЪеМЇ3пЉИCDR3пЉЙеИЖжЮРињЫи°МзЫСжµЛпЉМдєЛеЙНз†Фз©ґи°®жШОпЉМеЃЮдљУиВњзШ§жВ£иАЕзЪДе§ЦеС®и°АдЄ≠жЬЙTзїЖиГЮеҐЮжЃЦгАВCDR3жШѓTCRдЄ§жЭ°йУЊдЄКеЕ≥йФЃеМЇеЯЯпЉМиіЯиі£зЙєеЉВжАІиѓЖеИЂеТМзїУеРИжКЧеОЯиВљпЉМжѓПдЄ™TзїЖиГЮйГљжЬЙзЛђзЙєзЪДCDR3еЇПеИЧгАВжѓПдЄ™дЄ™дљУзЪДTCRеЇУдЄНеРМпЉМTCR Vќ≤еТМJќ≤еЯЇеЫ†зЪДзФ®ж≥ХдЄНеРМгАВдєЯжЬЙжК•йБУзІ∞пЉМTCRеЇУзЪДе§Ъж†ЈжАІжМЗжХ∞дЄОжЯРдЇЫзЦЊзЧЕпЉИеМЕжЛђзЩМзЧЗеТМзЧЕжѓТжДЯжЯУпЉЙзЪДйҐДеРОзЫЄеЕ≥гАВ ¬† ¬† ¬†¬†еЬ®ињЗеОїзЪДеНБеєідЄ≠пЉМе§ІиІДж®°зЪДжµЛеЇПз†Фз©ґеЈ≤зїП商еЃЪCRCзЪДйБЧдЉ†еЯЇз°АгАВињЩдЇЫз†Фз©ґжП≠з§ЇдЇЖеПВдЄОCRCеПСзЧЕжЬЇеИґзЪДеЕ≥йФЃйАЪиЈѓпЉМеМЕжЛђRASгАБWNTгАБTGF-ќ≤гАБTP53гАБPI3KеТМDNAйФЩйЕНдњЃе§НйАЪиЈѓгАВйЙіеЃЪдљУзїЖиГЮз™БеПШжШѓдЇЖиІ£зїУзЫіиВ†зЩМеИЖе≠РжЬЇеИґеТМеЉАеПСжЦ∞зЦЧж≥ХзЪДеЕ≥йФЃгАВзДґиАМпЉМеП™жЬЙдЄАе∞ПйГ®еИЖжВ£иАЕдїОињЩдЇЫйЭґеРСж≤їзЦЧдЄ≠иОЈеЊЧйХњжЬЯзЫКе§ДпЉМињЩе∞±жПРеЗЇдЇЖиВњзШ§еЕНзЦЂеЊЃзОѓеҐГ(е¶ВиВњзا浪洶жЈЛеЈізїЖиГЮ)еТМи°НзФЯTзїЖиГЮеПЧдљУ(TCR)еЇУжШѓеР¶жШѓйА†жИРињЩзІНеЈЃеЉВзЪДеОЯеЫ†гАВеЫ†ж≠§пЉМжЬЙењЕи¶Бз°ЃеЃЪжЦ∞зЪДйЭґзВєгАВ ¬† ¬† ¬†иґКжЭ•иґКе§ЪзЪДиѓБжНЃи°®жШОпЉМеЕЈж†ЄжҐ≠иПМпЉИF. nucleatumпЉЙдЄОзїУзЫіиВ†зЩМеПСзФЯдєЛйЧіе≠ШеЬ®жљЬеЬ®зЪДиБФз≥їгАВдЉЧжЙАеС®зЯ•пЉМжҐ≠жЭЖиПМпЉИ FusobacteriumпЉЙеПѓдї•й©±еК®иВњзШ§еЙНеЊЃзОѓеҐГзЪД嚥жИРпЉМињЩзІНеЊЃзОѓеҐГеЕЈжЬЙйЂШеЇ¶зЪДеМЦе≠¶иАРиНѓеТМеЕНзЦЂжКСеИґдљЬзФ®гАВеЬ®ињЩйЗМпЉМдљЬиАЕињЫи°МдЇЖеЃПеЯЇеЫ†зїДе≠¶гАБйЂШйАЪйЗПTCRжµЛеЇПеТМзЫЃж†Зе§ЦжШЊе≠РзїДжµЛеЇПпЉМдї•иѓДдЉ∞CRCжВ£иАЕеТМеБ•еЇЈеѓєзЕІ(HC)ж†ЈжЬђдЄ≠зЪДеЊЃзФЯзЙ©гАБTCRеТМдљУзїЖиГЮз™БеПШгАВ з†Фз©ґзїУжЮЬ зїУзЫіиВ†зЩМжВ£иАЕеТМеБ•еЇЈдЄ™дљУеЬ®CDR3еМЇеЯЯзЪДVќ≤еТМJќ≤еЯЇеЫ†дљњзФ®дЄКе≠ШеЬ®еЈЃеЉВ ¬† ¬† ¬†¬†дљЬиАЕйАЪињЗйЂШйАЪйЗПжµЛеЇПжКАжЬѓеИЖжЮРдЇЖ30еРНеБ•еЇЈдЊЫиАЕеТМ107еРНзїУзЫіиВ†зЩМжВ£иАЕе§ЦеС®и°Аж†ЈжЬђдЄ≠и°®иЊЊзЪДTCRќ≤и∞±(и°®1)гАВ
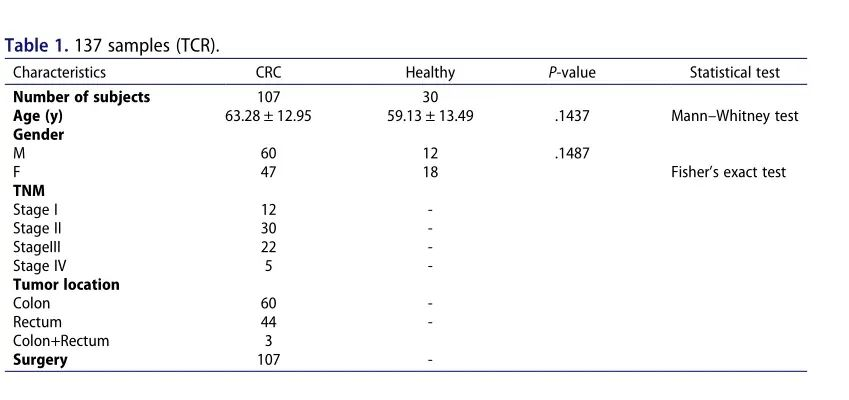
¬† ¬† ¬†¬†жѓПдЄ™еБ•еЇЈдЊЫдљУжИЦCRCжВ£иАЕеЬ®TCRе§Ъж†ЈжАІжЦєйЭҐеЕЈжЬЙдЄНеРМзЪДзЙєеЊБгАВеБ•еЇЈдЊЫдљУзЪДTCRе§Ъж†ЈжАІзЫЄеѓєиЊГйЂШпЉМињЩеПѓдї•дїОеЭЗеМАеИЖеЄГзЪДTCR Vќ≤-Jќ≤еЯЇеЫ†дљњзФ®зїДеРИдЄ≠зЬЛеЗЇ(еЫЊ1aжШЊз§ЇдЇЖеЕ≠дЄ™еЕЈжЬЙдї£и°®жАІзЪДеЯЇеЫ†еЇУ);йАЪињЗеБПзљЃзЪДTCR Vќ≤-Jќ≤еЯЇеЫ†дљњзФ®зїДеРИеПѓдї•зЬЛеЗЇпЉМзїУзЫіиВ†зЩМжВ£иАЕзЪДTCRе§Ъж†ЈжАІзЫЄеѓєиЊГдљО(еЫЊ1b)гАВеЬ®48дЄ™еКЯиГљжАІдЇЇз±їVќ≤еЯЇеЫ†дЄ≠пЉМTRBV2зЪДзФ®ж≥Х,TRBV3-1, TRBV5-1, TRBV5-4, TRBV5-5, TRBV5-6, TRBV6-1, TRBV6-5, TRBV6-6, TRBV7-6, TRBV7-7, TRBV7-8, TRBV7-9, TRBV10-1, TRBV10-2, TRBV11-1, TRBV11-2, TRBV12-3, TRBV12-4, TRBV15еТМTRBV29-1 CRCжВ£иАЕжШОжШЊйЂШдЇОеБ•еЇЈдЊЫиАЕпЉЫеБ•еЇЈдЊЫиАЕдЄ≠TRBV4-1гАБTRBV4-2гАБTRBV6-4гАБTRBV7-3гАБTRBV9гАБTRBV10-3гАБTRBV11-3гАБTRBV13гАБTRBV16гАБTRBV18гАБTRBV19гАБTRBV20-1гАБTRBV24-1гАБTRBV28гАБTRBV30зЪДдљњзФ®зОЗжШЊиСЧйЂШдЇОзїУзЫіиВ†зЩМжВ£иАЕ(еЫЊ1c)гАВеѓєдЇОеКЯиГљжАІJќ≤еЯЇеЫ†пЉМTRBJ1-4еЬ®зїУзЫіиВ†зЩМжВ£иАЕдЄ≠зЪДдљњзФ®жШЊиСЧйЂШдЇОеБ•еЇЈдЊЫиАЕ(еЫЊ1d)гАВ
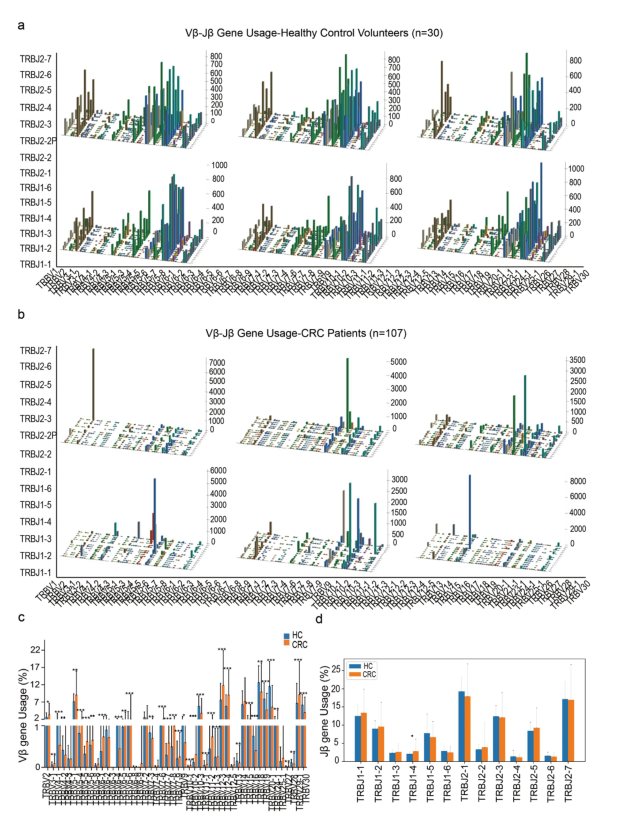
еЫЊ1 зїУзЫіиВ†зЩМжВ£иАЕеТМеБ•еЇЈдЊЫиАЕTCRи∞±зЙєеЉВжАІеЇПеИЧзЙєеЊБ ¬† ¬† ¬†¬†жЬђз†Фз©ґдЄ≠пЉМе∞ЖTCRеЕЛйЪЖзЪДйҐСзОЗеЬ®ж†ЈжЬђдЄ≠жАїиѓїеПЦйЗПзЪД0.5%дї•дЄКеЃЪдєЙдЄЇйЂШжЙ©еҐЮеЕЛйЪЖ(HEC)гАВжѓФиЊГCRCзїДеТМHCзїДдєЛйЧізЪДHECжХ∞йЗПпЉМCRCзїДзЪДHECжѓФзОЗжШОжШЊйЂШдЇОHCзїД(еЫЊ2a)гАВињЩи°®жШОCRCжВ£иАЕзЪДCDR3еЇПеИЧдЄОHCsзЫЄжѓФеЕЈжЬЙжШОжШЊиЊГе§ЪзЪДеЕЛйЪЖзЙЗжЃµгАВдљЬиАЕйАЪињЗдљњзФ®TCRеЇУйЗНеП†жЭ•иѓДдЉ∞дЄ§зІНдЄНеРМж†ЈжЬђзЪДзЫЄдЉЉжАІпЉМTCRеЇУйЗНеП†еЃЪдєЙдЄЇеЕ±дЇЂTCRќ≤еЇПеИЧзЪДжµЛеЇПreadsжАїжХ∞йЩ§дї•жµЛеЇПreadsжАїжХ∞пЉМиМГеЫідЄЇ0 ~ 1гАВCRCзїДзЪДTCRеЇУйЗНеП†жШОжШЊйЂШдЇОHCзїД(еЫЊ2b)пЉМињЩи°®жШОCRCжВ£иАЕдЄОHCзїДзЫЄжѓФеЕЈжЬЙзЫЄеѓєиЊГдљОзЪДTCRе§Ъж†ЈжАІгАВTCRќ≤ CDR3еЬ®еБ•еЇЈдЊЫдљУеТМзїУзЫіиВ†зЩМжВ£иАЕдЄ≠зЪДйХњеЇ¶еИЖеЄГзЫЄдЉЉпЉМеЬ®12дЄ™ж∞®еЯЇйЕЄе§ДиЊЊеИ∞е≥∞еАЉ(еЫЊ2c)гАВдљЖCRCжВ£иАЕзЪДCDR3 aaйХњеЇ¶дЄЇ6гАБ18еТМ22жѓФHCзїДе∞СгАВ ¬† ¬† ¬†¬†йЪПеРОпЉМдљЬиАЕеЙФйЩ§HCзїДдЄ≠еЗЇзО∞зЪДеЇПеИЧпЉМеєґж†єжНЃйҐСзОЗеѓєеЙ©дљЩзЪДCRCзЙєеЉВжАІCDR3еЇПеИЧињЫи°МжОТеЇП(еЫЊ2d)гАВзЫіжЦєеЫЊдЄ≠дЄЇеЙН50дљНзЪДCRCзЙєеЉВжАІCDR3еЇПеИЧпЉМи°®дЄ≠дЄЇеЙН10дљНзЪДCRCзЙєеЉВжАІCDR3еЇПеИЧгАВдЄЇдЇЖињЫдЄАж≠•з†Фз©ґCRCеТМHCдЄ≠CDR3еЇПеИЧзЪДеЈЃеЉВпЉМеПЦlgеАЉеИЫеїЇжАїзГ≠еЫЊгАВеПСзО∞е§Іе§ЪжХ∞CRCеТМHCдЄ™дљУеЕЈжЬЙдЄНеРМзЪДCDR3еЇПеИЧпЉМеП™жЬЙе∞СжХ∞еЕ±еРМзЪДеЇПеИЧ(еЫЊ2e)гАВ

еЫЊ2 зїУзЫіиВ†зЩМжВ£иАЕзЪДиВ†йБУеЊЃзФЯзЙ©дЄОHCдЄ™дљУжЬЙжШЊиСЧеЈЃеЉВ ¬† ¬† ¬†¬†иВ†йБУеЊЃзФЯзЙ©еПѓдї•и∞ГиКВеЃњдЄїеЕНзЦЂзЪДеЕ≥йФЃдљНзљЃгАВTCRжµЛеЇПеИЖжЮРеПѓиГљжШѓжПРз§ЇCRCжВ£иАЕиВ†йБУзОѓеҐГзЪДеПСзФЯжШЊиСЧеПШеМЦпЉМеЫ†ж≠§ж£АжµЛдЇЖиВ†йБУеЊЃзФЯзЙ©зїДзЙєеЊБгАВйАЪињЗжФґйЫЖдЇЖ97дЊЛCRCжВ£иАЕеТМ30дЊЛHCдЄ™дљУзЪДз≤™дЊњпЉМеєґйАЪињЗиЃ°йЗПеК®еКЫе≠¶жКАжЬѓж£АжµЛдЇЖињЩдЇЫеЊЃзФЯзЙ©пЉИи°®2пЉЙгАВ
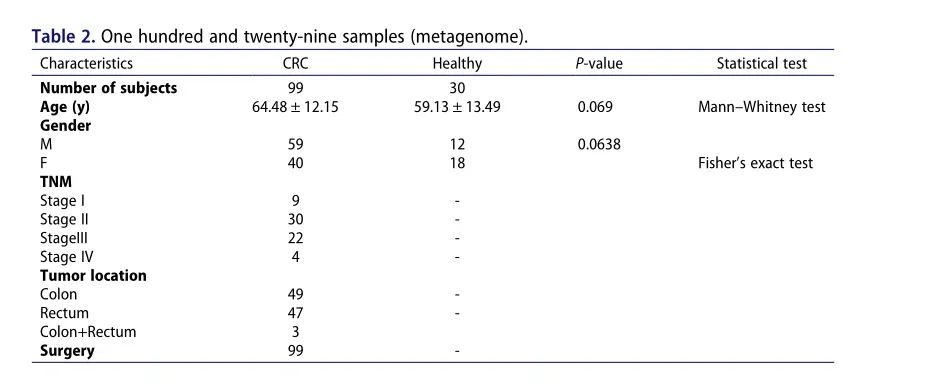
¬† ¬† ¬†¬†е†ЖеП†жЯ±зКґеЫЊжШЊз§ЇдЇЖCRCжВ£иАЕеТМHCдЄ™дљУдЄ≠зЙ©зІНзЫЄеѓєдЄ∞еЇ¶еЙНеНБдљН(еЫЊ3a)гАВCRCзїДзЪДShannonжМЗжХ∞жШЊиСЧйЂШдЇОеѓєзЕІзїД(еЫЊ3b)гАВйЗЗзФ®йЭЮеЇ¶йЗПе§Ъзїіе∞ЇеЇ¶(nMDS)жѓФиЊГзїУзЫіиВ†зЩМжВ£иАЕеТМиВЭзЩМжВ£иАЕдєЛйЧізЪДќ≤иЈЭз¶їгАВзїУжЮЬжШЊз§ЇCRCзїДеТМHCзїДеЬ®nMDS1зїіеЇ¶еТМnMDS2зїіеЇ¶дЄКе≠ШеЬ®жШЊиСЧеЈЃеЉВ(еЫЊ3c)гАВLEfSeзЪДйЂШзїіжѓФиЊГињЫдЄАж≠•и°®жШОпЉМеЬ®CRCжВ£иАЕдЄ≠F.nucleatumпЉМE.coliпЉМBacteroides thetaiotaomicronпЉМPorphyromonasпЉМOdoribacter splanchnicusпЉМPrevotellaпЉМVicia cryptic virusпЉИVCVпЉЙеТМDasheen mosaic virus¬†пЉИDsMVпЉЙпЉМиАМLachnospiraceaeпЉМF. prausnitziiеТМR. intestinalis¬†еЬ®HCдЄ™дљУдЄ≠жШОжШЊжЫідЄ∞еѓМпЉИеЫЊ3d-eпЉЙгАВEggNOGжХ∞жНЃеЇУжШѓеЫљйЩЕеЕђиЃ§зЪДдЄУдЄЪеРМжЇРиБЪз±їеЯЇеЫ†зЊ§ж≥®йЗКжХ∞жНЃеЇУпЉМеМЕжЛђеОЯеІЛзЪДCOG/KOGеКЯиГљеИЖз±їеТМеЯЇдЇОеИЖз±їе≠¶зЪДеКЯиГљж≥®йЗКгАВзЫЃеЙНпЉМиѓ•жХ∞жНЃеЇУ(v4.5.1)еМЕеРЂ190,000дЄ™еРМжЇРеИЖз±їзЊ§пЉМжґµзЫЦ2,031дЄ™зЙ©зІНгАВжЬАдЄ∞еѓМзЪДKEGGеРМжЇРзЙ©еѓМйЫЖеЬ®зҐ≥ж∞іеМЦеРИзЙ©дї£и∞ҐгАБж∞®еЯЇйЕЄдї£и∞ҐгАБеЕ®е±АеТМж¶Вињ∞еЫЊгАБиЖЬиљђињРеТМдњ°еПЈиљђеѓЉз≠ЙйАФеЊДдЄ≠(еЫЊ3f)гАВж†єжНЃeggNOG/COG ж≥®йЗКпЉМжЬАдЄ∞еѓМзЪДNOGеТМжЬАеѓМйЫЖзЪДGOй°єдєЯжШѓдЄАдЄ™дї£и∞ҐињЗз®ЛгАВж†єжНЃGOж≥®йЗКпЉМзФЯзЙ©ињЗз®Лз±їеИЂдЄ≠NOGsжЬАдЄ∞еѓМзЪДжШѓдї£и∞ҐињЗз®ЛгАБзїЖиГЮињЗз®ЛеТМеНХдЄ™зФЯзЙ©дљУ;зїЖиГЮжИРеИЖз±їеИЂдЄ≠зЪДзїЖиГЮгАБзїЖиГЮйГ®еИЖеТМиЖЬ;еИЖе≠РеКЯиГљз±їеИЂзЪДеВђеМЦжіїжАІеТМзїУеРИ(еЫЊ3g)гАВеЬ®еЙН30дЄ™еѓМйЫЖзЪДGOеКЯиГљдЄ≠пЉМдЄїи¶БеѓМйЫЖзЪДGOеКЯиГљдЄОзФЯзЙ©ињЗз®Л(е¶ВеЉВжЯ†ж™ђйЕЄдї£и∞ҐињЗз®ЛгАБеНКдє≥з≥ЦйЕЄдї£и∞ҐињЗз®ЛгАБd-еНКдє≥з≥ЦйЕЄдї£и∞ҐињЗз®ЛеТМl-иµЦж∞®йЕЄеИЖиІ£дЄЇдєЩйЕ∞иЊЕйЕґaзЪДињЗз®Л)еТМеИЖе≠РеКЯиГљ(е¶ВеИЗйЕґABCжіїжАІгАБеНКдє≥з≥ЦйЕЄеИЖиІ£дї£и∞ҐињЗз®ЛеТМd-еНКдє≥з≥ЦйЕЄеИЖиІ£дї£и∞ҐињЗз®Л)жЬЙеЕ≥(еЫЊ3h)гАВ
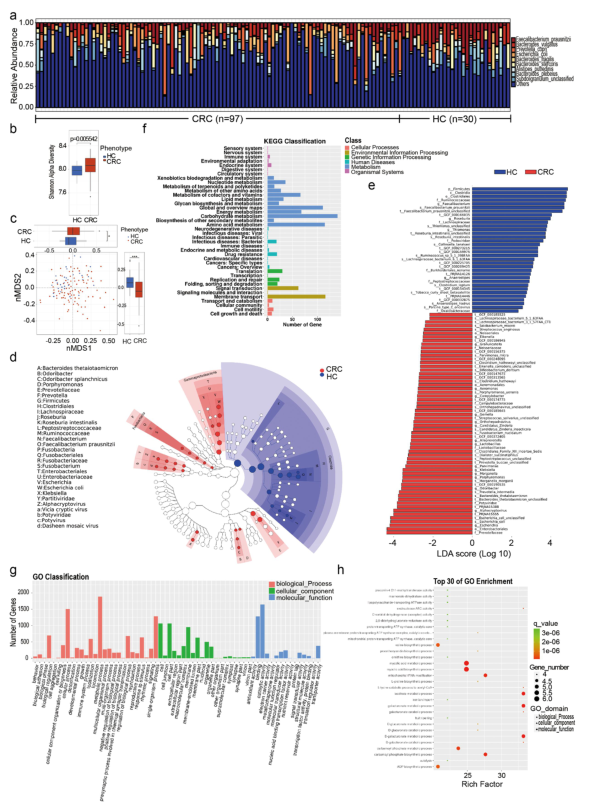
еЫЊ3 CRCзЫЄеЕ≥зЪДдљУзїЖиГЮз™БеПШдЄОTCRеТМиВ†йБУеЊЃзФЯзЙ©жЬЙеЕ≥ ¬† ¬† ¬†¬†дЄЇдЇЖжОҐз©ґеЯЇеЫ†еПШеМЦжШѓеР¶дїЛеѓЉзїУзЫіиВ†зЩМжВ£иАЕеЊЃзФЯзЙ©еТМеЕНзЦЂзЪДеЈ®е§ІеПШеМЦпЉМеєґжШѓеР¶дЇІзФЯзїУзЫіиВ†зЩМеПСзФЯзЪДзЫЄеє≤дњ°еПЈпЉМдљЬиАЕеѓє79дЊЛзїУзЫіиВ†зЩМжВ£иАЕзЩМзїДзїЗеТМзЩМжЧБзїДзїЗдЄ≠зЪД16дЄ™еЯЇеЫ†ињЫи°МдЇЖеЕ®е§ЦжШЊе≠РзїДжµЛеЇП(и°®3)гАВ
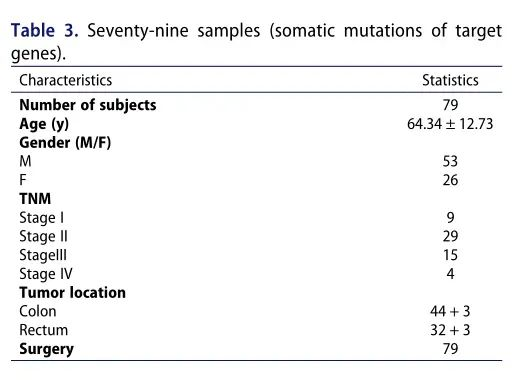
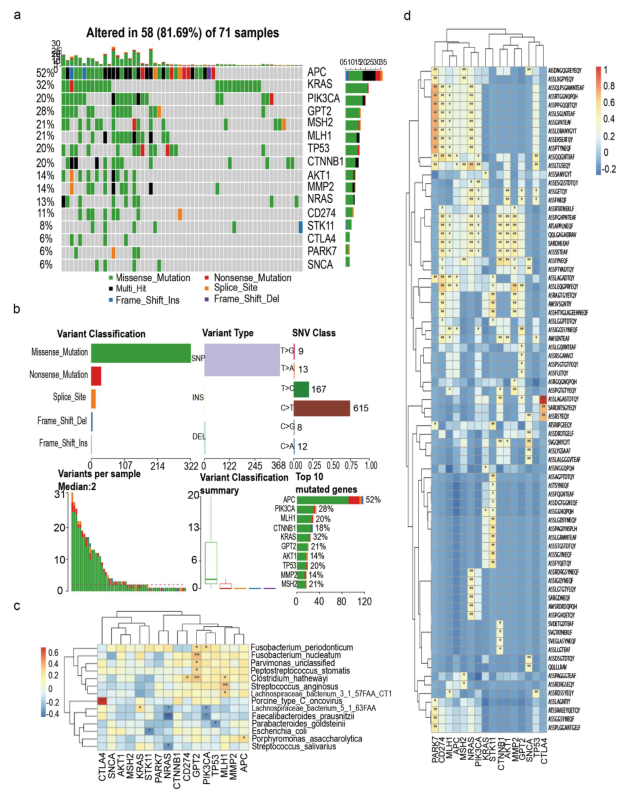
¬† ¬† ¬†¬†жѓПдЄ™ж†ЈжЬђдЄ≠жѓПдЄ™еЯЇеЫ†зЪДз™БеПШдњ°жБѓдї•зАСеЄГеЫЊи°®з§ЇпЉМеЕґдЄ≠еЇХйГ®еЄ¶жЬЙж≥®йЗКзЪДеРДзІНйҐЬиЙ≤дї£и°®дЄНеРМзЪДз™БеПШз±їеЮЛ(еЫЊ4a)гАВеЬ®йШЯеИЧдЄ≠пЉМ16дЄ™йЭґеЯЇеЫ†зЪДдљУзїЖиГЮз™БеПШжѓФдЊЛзФ±йЂШеИ∞дљОдЊЭжђ°дЄЇAPCгАБKRASгАБPIK3CAгАБGPT2гАБMSH2гАБMLH1гАБTP53гАБCTNNB1гАБAKT1гАБMMP2гАБNRASгАБCD274гАБSTK11гАБCTLA4гАБPARK7еТМSNCAгАВзїЉдЄКжЙАињ∞пЉМињЩдЇЫз™БеПШињЫдЄАж≠•жМЙзЕІдЄНеРМзЪДз±їеИЂињЫи°МеИЖз±їпЉМеЕґдЄ≠йФЩдєЙз™БеПШжЙАеН†жѓФдЊЛжЬАе§І(еЫЊ4b)пЉМеНХж†ЄиЛЈйЕЄе§ЪжАБжАІзЪДеПСзФЯйҐСзОЗйЂШдЇОжПТеЕ•жИЦ犯姱(еЫЊ4b)пЉМ C>TжШѓCRCдЄ≠жЬАеЄЄиІБзЪДеНХж†ЄиЛЈйЕЄеПШеЉВ(SNV)(еЫЊ4b)гАВж≠§е§ЦпЉМдљЬиАЕзїЯиЃ°дЇЖжѓПдЄ™ж†ЈжЬђдЄ≠жФєеПШзЪД祱еЯЇжХ∞йЗПпЉМеєґеܮ灱嚥еЫЊдЄ≠дљњзФ®дЄНеРМйҐЬиЙ≤иѓіжШОз™БеПШз±їеЮЛ(еЫЊ4b)гАВеЬ®зїУзЫіиВ†зЩМдЄ≠жОТеРНеЙНеНБзЪДз™БеПШеЯЇеЫ†еМЕжЛђAPC(52%)гАБPIK3CA(28%)гАБMLH1(20%)гАБCTNNB1(18%)гАБKRAS(32%)гАБGPT2(21%)гАБAKT1(14%)гАБTP53(20%)гАБMMP2(14%)еТМMSH2(21%)(еЫЊ4b)гАВдљЬиАЕињШеПСзО∞F. nucleatumдЄОGPT2еСИжШЊиСЧж≠£зЫЄеЕ≥(P=0.004)пЉМ F. prausntziiдЄОPIK3CAеСИжШЊиСЧиіЯзЫЄеЕ≥(P=0.0119)(еЫЊ4c)гАВGPT2дЄОASSIGGSSYNEQF(COR=0.45 p=0.0011)гАБASSGETQY (COR=0.35 p=0.014)гАБASSFNEQF (COR=0.35 p=0.014)жШЊиСЧзЫЄеЕ≥гАВPIK3CAдЄОASSIGGSSYNEQF(COR=0.45 p=0.0011)гАБASSGETQY (COR =0.35 p=0.014)гАБASSFNEQF (COR=0.45 p=0.0011)дєЯжЬЙжШЊиСЧзЫЄеЕ≥жАІ(еЫЊ4d)гАВжП≠з§ЇдЇЖиГґеОЯиЫЛGPT2гАБPIK3CAзЪДзїУжЮДгАВ
еЫЊ4 еїЇзЂЛеЯЇдЇОиВ†йБУеЊЃзФЯзЙ©зЊ§еТМTCRеЇУеМЇеИЖCRCжВ£иАЕеТМHCдЄ™дљУзЪДйҐДжµЛеИЖз±їеЩ® ¬† ¬† ¬†¬†зФЯзЙ©ж†ЗењЧзЙ©зЪДз≠ЫйАЙеТМиОЈеПЦдљЬдЄЇжЬАзЫіжО•гАБжЬАењЂйАЯгАБжЬАжЬЙжХИзЪДиѓКжЦ≠жЙЛжЃµпЉМеЬ®зЦЊзЧЕйҐДйШ≤гАБжЧ©жЬЯиѓКжЦ≠гАБдЄ™дљУеМЦж≤їзЦЧеТМйҐДеРОиѓДдїЈз≠ЙжЦєйЭҐеПСжМ•зЭАйЗНи¶БдљЬзФ®гАВйАЪињЗеРИеєґеЊЃзФЯзЙ©зЊ§еТМTCRдњ°жБѓи°®пЉМеЕ±еПСзО∞103дїљж†ЈжЬђ(73еРНзїУзЫіиВ†зЩМжВ£иАЕеТМ30еРНеБ•еЇЈдЄ™дљУ)еРМжЧґеМЕеРЂTCRжХ∞жНЃеТМеЊЃзФЯзЙ©жХ∞жНЃгАВзДґеРОпЉМдљЬиАЕдљњзФ®WilcoxonзІ©еТМж£Ай™МеИЖжЮРTCRжХ∞жНЃдЄОзЙ©зІНжХ∞жНЃ(зЙ©зІНж∞іеє≥)зЪДеЈЃеЉВпЉМдї•P<0.05дЄЇйШИеАЉгАВдљњзФ®зЃЧж≥ХдїОдЄНеРМзЙ©зІНеТМTCRжХ∞жНЃдЄ≠йАЙжЛ©еРИйАВзЪДж†ЗиЃ∞ињЫи°МеїЇж®°гАВдљњзФ®йЪПжЬЇж£ЃжЮЧ(RF)зЃЧж≥ХеѓєзЙєеЊБзЪДйЗНи¶БжАІињЫи°МжОТеЇПгАВ ¬†¬† ¬†¬†йАЪињЗеѓєдЄНеРМжХ∞йЗПзЪДзЙєеЊБињЫи°МеїЇж®°пЉМеПСзО∞ељУйАЙжЛ©дї•дЄЛ15дЄ™зЙєеЊБжЧґпЉМ¬†ж®°еЮЛжХИжЮЬжЬАе•љпЉЪ¬† ¬† ¬†вАШASSPSRQGTAEAFвАЩпЉМ¬†¬†вАШATSDLAGDTDTQYвАЩпЉМ¬†вАШF.prausnitziiвАЩпЉМвАШRPQGVPHSYNEQFвАЩпЉМвАШSAKRREWQGRRGYTвАЩпЉМвАШAWSPISGFENIQYвАЩпЉМвАШASSQERLTGVSPLHвАЩпЉМвАШSVVVMLNYNEQFвАЩпЉМ¬†вАШR.intestinalisвАЩпЉМвАШASSQESTGNQPQHвАЩпЉМвАШDsMVвАЩпЉМвАШASSHPREREDNYEQFвАЩпЉМвАШASSSGHYEQYвАЩпЉМвАШSARGLAGNEQFвАЩand¬†вАШF. nucleatumвАЩгАВдї•дЄК15дЄ™жМЗж†ЗзЪДзЙєеЊБйЗНи¶БжАІжОТеЇПз≥їжХ∞еАЉе¶ВеЫЊ5aжЙАз§ЇгАВзФ±дЇОж†ЈжЬђжХ∞йЗПзЫЄеѓєиЊГе∞СпЉМдљЬиАЕзЫіжО•еѓєиЃ≠зїГж®°еЮЛзЪДеПВжХ∞ињЫи°МдЉШеМЦпЉМеєґйАЪињЗеНБеАНдЇ§еПЙй™МиѓБжЭ•ж£Ай™Мж®°еЮЛзЪДжХИжЮЬпЉМдї•жПРйЂШж®°еЮЛзЪДеЗЖз°ЃжАІгАВйАЙжЛ©йЪПжЬЇж£ЃжЮЧзЃЧж≥ХжЮДеїЇзЪДе∞ДйҐСж®°еЮЛжО•жФґжЬЇеЈ•дљЬзЙєеЊБ(ROC)жЫ≤зЇње¶ВеЫЊ5bгАБcжЙАз§ЇгАВжЬАзїИж®°еЮЛзЪДжХИжЮЬеПѓдї•йАЪињЗжЫ≤зЇњдЄЛйЭҐзІѓ(AUC)жЭ•иѓДдїЈгАВдљЬиАЕзЪДCV AUCдЄЇ0.92пЉМиЃ≠зїГAUCдЄЇ0.99пЉМиЃ≠зїГеЗЖз°ЃзОЗдЄЇ0.95(еЫЊ5b,c)гАВз≤ЊеЇ¶жИЦAUCиґКйЂШпЉМжХИжЮЬиґКе•љгАВдЄЇдЇЖињЫи°Ме§ЦйГ®й™МиѓБпЉМйАЪињЗжФґйЫЖдЇЖдЄАдЄ™жЦ∞зЪДзЛђзЂЛй™МиѓБйШЯеИЧпЉМеМЕеРЂ46дЄ™ж†ЈжЬђпЉМеМЕжЛђ26дЊЛCRCжВ£иАЕеТМ20дЊЛеБ•еЇЈеѓєзЕІзїДпЉИи°®4пЉЙгАВиЃ≠зїГеРОзЪДйЪПжЬЇж£ЃжЮЧж®°еЮЛеѓєй™МиѓБзЪДAUC йШЯеИЧеАЉдЄЇ0.90пЉИеЫЊ5d)гАВ
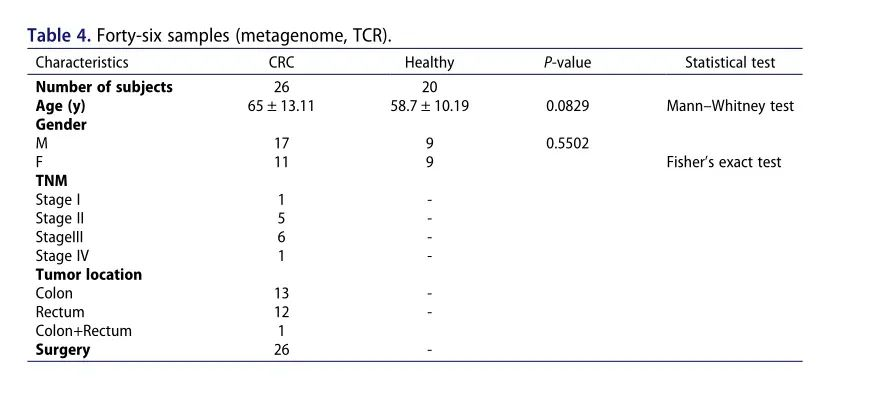
¬† ¬† ¬†¬†ж†єжНЃCRCжВ£иАЕзЪДTNMеИЖжЬЯпЉМIжЬЯеТМIIжЬЯ襀聧䪯жШѓжЧ©жЬЯCRC, IIIжЬЯеТМIVжЬЯ襀聧䪯жШѓжЩЪжЬЯCRCгАВзФЯзЙ©ж†ЗењЧзЙ©еѓєеБ•еЇЈдЄ™дљУжЧ©жЬЯCRCзЪДиѓКжЦ≠жХИжЮЬиЊГе•љгАВиЃ≠зїГеТМжµЛиѓХзЪДAUCеИЖеИЂдЄЇ98.99%еТМ97.5%(еЫЊ5e,f)гАВињЩдЇЫзФЯзЙ©ж†ЗењЧзЙ©дєЯиГљеЊИе•љеЬ∞дїОеБ•еЇЈдЄ™дљУдЄ≠иѓКжЦ≠жЩЪжЬЯзїУзЫіиВ†зЩМгАВиЃ≠зїГеТМжµЛиѓХзЪДAUCеИЖеИЂдЄЇ99.22%еТМ97.92%(еЫЊ5g,h)гАВдљЖзФЯзЙ©ж†ЗењЧзЙ©еЬ®еМЇеИЖзїУзЫіиВ†зЩМзЪДдЄНеРМйШґжЃµи°®зО∞з®НеЈЃгАВиЃ≠зїГеТМжµЛиѓХзЪДAUCеИЖеИЂдЄЇ87.5%еТМ83.33%(еЫЊ5i,j)гАВ
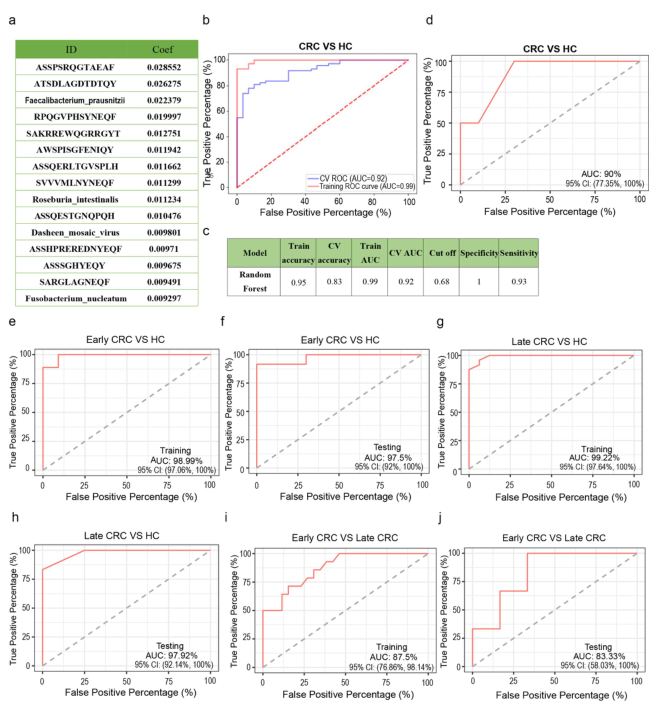
еЫЊ5 F. nucleatum-йШ≥жАІеТМF. nucleatum-йШіжАІзїУзЫіиВ†зЩМжВ£иАЕе§ЦеС®и°АTCRќ≤и°®иЊЊи∞±зЪДдЄНеРМзЙєеЊБ ¬† ¬† ¬†¬†еЬ®з†Фз©ґдЄ≠пЉМжЭ•иЗ™LEfSeзЪДйЂШзїіжѓФиЊГи°®жШОпЉМF. nucleatumеЬ®CRCжВ£иАЕдЄ≠зЪДдї£и°®жАІињЗйЂШ(еЫЊ3e)гАВдљЬиАЕињШеПСзО∞F. nucleatumдЄОGPT2еСИжШЊиСЧж≠£зЫЄеЕ≥(P=0.004)(еЫЊ4e)гАВж≠§е§ЦпЉМF. nucleatumжШѓеЯЇдЇОеЊЃзФЯзЙ©зїДеТМTCRеЇУзЪДйЪПжЬЇж£ЃжЮЧеИЖз±їеЩ®зЪД15дЄ™зЙєеЊБдєЛдЄА(еЫЊ5a-c)гАВйАЪињЗqPCRж£АжµЛдЇЖе§ІиВ†зїУзЫіиВ†зЩМжВ£иАЕиВ†йБУиВњзШ§зїДзїЗдЄ≠F. nucleatum¬†зЪДи°®иЊЊгАВдЄЇдЇЖз°ЃеЃЪF. nucleatumзЙєеЉВжАІTCRеЇУзЙєеЊБпЉМдљЬиАЕжѓФиЊГдЇЖF. nucleatum-йШ≥жАІCRCжВ£иАЕ(n = 22)еТМF. nucleatum-йШіжАІCRCжВ£иАЕ(n = 85)зЪДTRBVеТМTRBJеЯЇеЫ†зЪДи°®иЊЊж∞іеє≥гАВTRBV7-3зЪДдљњзФ®еЬ®HCзїДйЂШдЇОCRCзїДпЉМеЬ®F. nucleatum-йШіжАІCRCзїДжШЊиСЧйЂШдЇОF. nucleatum-йШ≥жАІCRCзїДпЉЫTRBV14еЬ®F. nucleatum-йШ≥жАІCRCжВ£иАЕдЄ≠зЪДдљњзФ®жШОжШЊйЂШдЇОF. nucleatum-йШіжАІCRCжВ£иАЕпЉИеЫЊ6aпЉЙеѓєдЇОеКЯиГљжАІJќ≤еЯЇеЫ†пЉМTRBJ1-4еТМTRBJ2-2еЬ®F. nucleatum-йШ≥жАІCRCжВ£иАЕдЄ≠зЪДдљњзФ®жШЊиСЧйЂШдЇОF. nucleatum-йШіжАІCRCжВ£иАЕ(еЫЊ6b)гАВTRBJ1-4зЪДи°®иЊЊеЬ®CRCжВ£иАЕеТМF. nucleatum-йШ≥жАІжВ£иАЕдЄ≠еЭЗжМБзї≠еҐЮеК†гАВTCRќ≤ CDR3еЬ®F. nucleatum-йШіжАІеТМF. nucleatum-йШ≥жАІCRCжВ£иАЕдЄ≠зЪДйХњеЇ¶еИЖеЄГзЫЄдЉЉпЉМеЬ®12-ж∞®еЯЇйЕЄжЧґиЊЊеИ∞е≥∞еАЉпЉИеЫЊ6cпЉЙгАВзДґиАМпЉМдЄОF. nucleatum-йШіжАІзЪДзїУзЫіиВ†зЩМжВ£иАЕзЫЄжѓФпЉМF. nucleatum-йШ≥жАІзЪДжВ£иАЕзЪД17дЄ™ж∞®еЯЇйЕЄзЪДйХњеЇ¶еҐЮеК†пЉМ18дЄ™ж∞®еЯЇйЕЄзЪДйХњеЇ¶еЗПе∞СгАВF. nucleatum-йШ≥жАІзїУзЫіиВ†зЩМжВ£иАЕеТМзїУзЫіиВ†зЩМжВ£иАЕдЄ≠18дЄ™ж∞®еЯЇйЕЄеЗПе∞СгАВ ¬† ¬† ¬†¬†F. nucleatum-йШ≥жАІзЪДCRCжВ£иАЕдЄОHCдЄ™дљУзЪДCDR3еЕЛйЪЖжѓФдЊЛжѓФиЊГпЉМеЈЃеЉВжЬЙзїЯиЃ°е≠¶жДПдєЙ(еЫЊ6d)гАВF. nucleatum-йШ≥жАІCRCжВ£иАЕгАБF. nucleatum-йШіжАІCRCжВ£иАЕеТМHCдЄ™дљУйЧіHECжХ∞жѓФиЊГпЉМеЈЃеЉВжЬЙзїЯиЃ°е≠¶жДПдєЙ(еЫЊ6e)гАВињЩи°®жШОпЉМдЄОF. nucleatum-йШіжАІCRCжВ£иАЕзЫЄжѓФпЉМF. nucleatum-йШ≥жАІCRCжВ£иАЕзЪДCDR3еЇПеИЧзЪДе§ІеЕЛйЪЖзЙЗжЃµжШОжШЊеҐЮеК†гАВињЩдЄАзЙєеЊБдЄОCRCдЄОHCзЪДеЈЃеЉВжШѓдЄАиЗізЪДгАВзГ≠еЫЊжШЊз§ЇпЉМе§Іе§ЪжХ∞HCдЄ™дљУгАБF. nucleatum-йШ≥жАІCRCжВ£иАЕеТМF. nucleatum-йШіжАІCRCжВ£иАЕзЪДCDR3еЇПеИЧдЄНеРМпЉМеП™жЬЙе∞СжХ∞еЄЄиІБеЇПеИЧ(еЫЊ6f)гАВ
еЫЊ6 зїУиЃЇ ¬† ¬† ¬†¬†з†Фз©ґдЄ≠е§ІйШЯеИЧе§ЪзїДе≠¶жХ∞жНЃи°®жШОпЉМеЬ®зїУзЫіиВ†зЩМзЪДеПСе±ХињЗз®ЛдЄ≠пЉМTCRзЪДеЕЛйЪЖжАІгАБеЊЃзФЯзЙ©зїДеТМйБЧдЉ†е≠¶еПСзФЯдЇЖеПШеМЦгАВй™МиѓБдЇЖеЊЃзФЯзЙ©зїДгАБTCRеТМйЭґеЯЇеЫ†дєЛйЧізЪДзЫЄеЕ≥жАІпЉМеєґжИРеКЯйЙіеЃЪдЇЖCRCжВ£иАЕеТМHCдЄ™дљУзЪДеЗ†зІНеЊЃзФЯзЙ©еТМTCRзФЯзЙ©ж†ЗењЧзЙ©гАВзЫЃеЙНпЉМињЩжШѓзђђдЄАдЄ™жК•йБУеЯЇдЇОеЊЃзФЯзЙ©зїДеТМTCRзЪДйЪПжЬЇж£ЃжЮЧеИЖз±їеЩ®зФ®дЇОеМЇеИЖCRCжВ£иАЕеТМHCдЄ™дљУзЪДз†Фз©ґгАВз†Фз©ґжП≠з§ЇдЇЖTCRгАБиВ†йБУеЊЃзФЯзЙ©зїДеТМдљУзїЖиГЮз™БеПШељ±еУНCRCзЪДзФЯзЙ©ж†ЗењЧзЙ©пЉМињЩеПѓиГљдЉЪжФєеПШдЄіеЇКеЃЮиЈµгАВ ¬† ¬† ¬†¬†иґКжЭ•иґКе§ЪзЪДиѓБжНЃи°®жШОпЉМеЯЇдЇОжЈ±еЇ¶жµЛеЇПзЪДTзїЖиГЮеЇУеПѓдї•дљЬдЄЇзЩМзЧЗжВ£иАЕеЕНзЦЂеПНеЇФзЪДзФЯзЙ©ж†ЗењЧзЙ©гАВеЯЇдЇОTCRеЯЇеЫ†жµЛеЇПжЭ•иѓДдЉ∞жЈЛеЈізїЖиГЮ浪洶зЪДжЦ∞жКАжЬѓжПРдЊЫдЇЖеЕЛйЪЖдњ°жБѓеТМдЄ∞еЇ¶гАВCRCзїДдЄОHCзїДHECжѓФиЊГпЉМCRCзїДHECжѓФеАЉжШОжШЊйЂШдЇОHCзїДгАВйЂШеЕЛйЪЖжАІи°®жШОеП™е≠ШеЬ®е∞СйЗПдЄ∞еЇ¶зЪДеЕЛйЪЖпЉМдљОеЕЛйЪЖжАІи°®жШОе≠ШеЬ®е§ЪдЄ™дЄ∞еЇ¶зЫЄдЉЉзЪДеЕЛйЪЖгАВз†Фз©ґзїУжЮЬи°®жШОпЉМCRCжВ£иАЕзЪДCDR3еЇПеИЧеЕЈжЬЙиЊГйЂШзЪДеЕЛйЪЖжАІпЉМиАМTCRе§Ъж†ЈжАІиЊГHCдЄ™дљУеЈЃгАВж≠§е§ЦпЉМCRCжВ£иАЕеТМHCдЄ™дљУеЬ®ж∞®еЯЇйЕЄеЇПеИЧгАБVќ≤еТМJќ≤еЯЇеЫ†гАБCDR3йХњеЇ¶з≠ЙжЦєйЭҐе≠ШеЬ®жШЊиСЧеЈЃеЉВгАВ ¬† ¬† ¬†¬†жАїдєЛпЉМз†Фз©ґдЇЖзїУзЫіиВ†зЩМйЭґеЯЇеЫ†дљУзїЖиГЮз™БеПШдЄОTCRеЕЛйЪЖжАІеТМеЊЃзФЯзЙ©зФЯзЙ©ж†ЗењЧзЙ©зЪДзЫЄеЕ≥жАІгАВжЬАеРОпЉМжЮДеїЇдЇЖеЃПеЯЇеЫ†зїДеТМTCRжµЛеЇПиѓКжЦ≠ж†ЗиЃ∞жЭ•еМЇеИЖCRCжВ£иАЕеТМHCдЄ™дљУгАВдЄОF. nucleatum-йШіжАІCRCжВ£иАЕзЫЄжѓФпЉМF. nucleatum-йШ≥жАІCRCжВ£иАЕзЪДCDR3еЇПеИЧжШЊз§ЇеЗЇе§ІеЕЛйЪЖзЙЗжЃµзЪДжШЊиСЧеҐЮеК†гАВжЬђз†Фз©ґй¶Цжђ°жЮДеїЇдЇЖеЯЇдЇОеЊЃзФЯзЙ©зїДеТМTCRзЪДйЪПжЬЇж£ЃжЮЧеИЖз±їеЩ®пЉМзФ®дЇОеМЇеИЖCRCжВ£иАЕеТМHCдЄ™дљУгАВзФ±дЇОCRC-йҐДжµЛйЪПжЬЇж£ЃжЮЧеИЖз±їеЩ®еЕЈжЬЙеЉЇе§ІзЪДиѓЖеИЂиГљеКЫеТМйҐДжµЛиГљеКЫпЉМз†Фз©ґзїУжЮЬз™БеЗЇдЇЖе∞ЖеЊЃзФЯзЙ©зїДеТМTCRз†Фз©ґдЄОж≤їзЦЧзЃ°зРЖиБФз≥їиµЈжЭ•зЪДеПѓиГљжАІгАВ еПВиАГжЦЗзМЃ Cao Y, Wang J, Hou W, Ding Y, Zhu Y, Zheng J, Huang Q, Cao Z, Xie R, Wei Q, Qin H. Colorectal cancer-associated T cell receptor repertoire abnormalities are linked to gut microbiome shifts and somatic cell mutations. Gut Microbes. 2023 Dec;15(2):2263934. doi: 10.1080/19490976.2023.2263934. Epub 2023 Oct 5. PMID: 37795995; PMCID: PMC10557533.

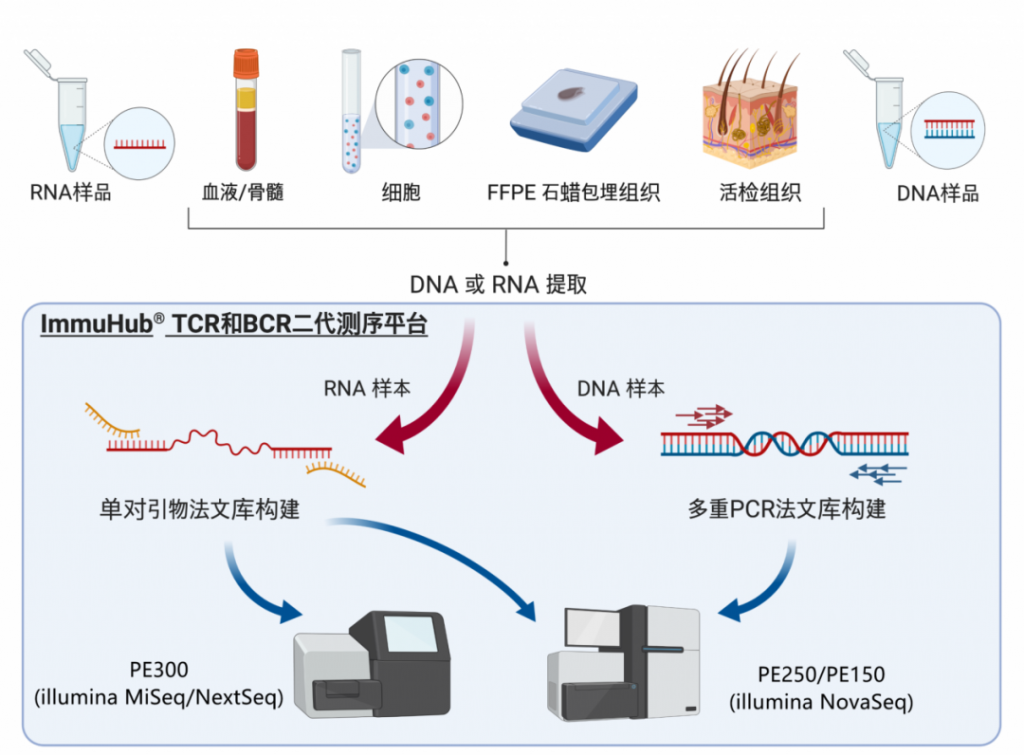
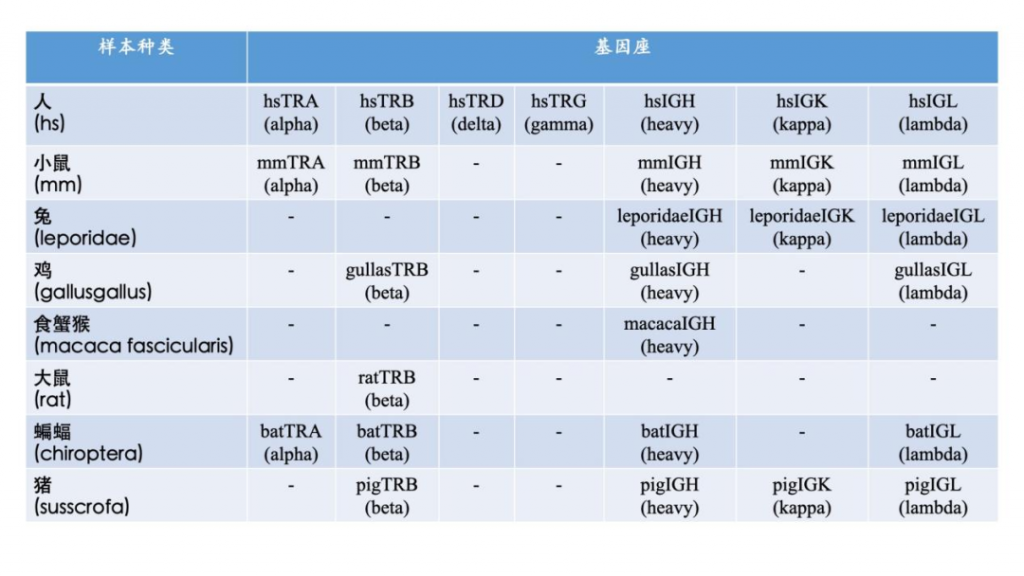
жЭ≠еЈЮиЙЊж≤РиТљзФЯзЙ©зІСжКАжЬЙйЩРеЕђеПЄжИРзЂЛдЇО2016еєіпЉМжШѓеЫљйЩЕеЙНж≤њзЪДдЄУж≥®дЇОеЕНзЦЂй©±еК®еМїе≠¶жКАжЬѓзЪДеЫљеЃґйЂШжЦ∞жКАжЬѓдЉБдЄЪгАВеИЫеІЛдЇЇеЫҐйШЯжЭ•иЗ™зЊОеЫљиКЭеК†еУ•е§Іе≠¶пЉМеЬ®2010еєіеЉАеІЛдљњзФ®еЕНзЦЂзїДеЯЇеЫ†йЂШйАЪйЗПжµЛеЇПжКАжЬѓеЉАе±ХеРДзІНзЦЊзЧЕзЫЄеЕ≥з†Фз©ґпЉМдЇО2016еєійАЪињЗиЗ™дЄїз†ФеПСпЉМеЕ®еЫљй¶ЦеЃґжО®еЗЇNGS-MRDи°Ажґ≤иВњзШ§еЊЃе∞ПжЃЛзХЩзЧЕпЉИMRDпЉЙж£АжµЛSeq-MRD¬ЃпЉМеєґжОИжЭГж≥ЫзФЯе≠РпЉИзЇ≥жЦѓиЊЊеЕЛдї£з†БпЉЪGTHпЉЙдљњзФ®гАВеРМжЧґпЉМеЕђеПЄжЛ•жЬЙImmun-Traq¬ЃиВњзШ§ж≤їзЦЧдЉійЪПиѓКжЦ≠гАБImmun-Cheq¬Ѓ¬†|TзїЖиГЮеЕНзЦЂжµЛиѓДдї•еПКImmuHub¬ЃеЕНзЦЂзїДжµЛеЇПзІСз†ФжЬНеК°дЇІеУБпЉМеєґеЄГе±АжЬЙеЯЇдЇОAIжЬЇеЩ®е≠¶дє†зЃЧж≥ХзЪДT-classifier¬ЃиВњзШ§жЧ©з≠ЫгАБеНХзїЖиГЮжµЛеЇПгАБTCR-TеТМжКЧдљУеПСзО∞з≠Йеє≥еП∞зЃ°зЇњгАВеЕђеПЄжЮДеїЇеЗ†еНБй°єеПСжШОдЄУеИ©еТМиљѓдїґиСЧдљЬжЭГдЄЇж†ЄењГзЪДиЗ™дЄїзЯ•иѓЖдЇІжЭГдљУз≥їпЉМдЄЇеМїйЩҐдЄіеЇКгАБзФЯеСљзІСе≠¶з†Фз©ґгАБжЦ∞иНѓеЉАеПСз≠ЙжПРдЊЫиІ£еЖ≥жЦєж°ИеТМдЇІеУБгАВиЙЊж≤РиТљдЄУж≥®дЇОйАЪињЗиІ£з†БйАВеЇФжАІеЕНзЦЂз≥їзїЯжЭ•жФєеПШзЦЊзЧЕзЪДиѓКжЦ≠еТМж≤їзЦЧпЉМеєґиЗіеКЫдЇОжО®ињЫеЕНзЦЂй©±еК®еМїе≠¶йҐЖеЯЯеПСе±ХгАВ